|
Выпуск 272 (English/Russian)
Лаборатория Наномир
Когда реальность открывает тайны,
уходят в тень и
меркнут чудеса ... Auto translation: When the reality reveals secrets, Miracles fade into the background and grow dim... Проверка: Когда действительность раскрывает секреты, Чудеса исчезают в фон и становятся тусклыми... Стандарт белковых структур (проект) PIKO standard (project) Introduction. METHOD Method description
METHOD The strong correlation dependence of spatial structure
METHOD of the protein from its nucleotide sequence was
METHOD theoretically predicted by physical modelling,
METHOD experimentally discovered and statistically confirmed.
METHOD In the process of biosynthesis the third nucleotide of
METHOD the codon controls the orientation of the amino acid
METHOD forming the concrete spatial isomer that is the
METHOD conformation of the protein molecule cutting off
METHOD competition
ways of the forming of 2D and 3D structures.
METHOD On this base the computer program "Pikotechnology" for
METHOD the prediction
of 2D structure of the protins on their
METHOD nucleotide sequence was created. Composition genetic code table (1992) Samples:
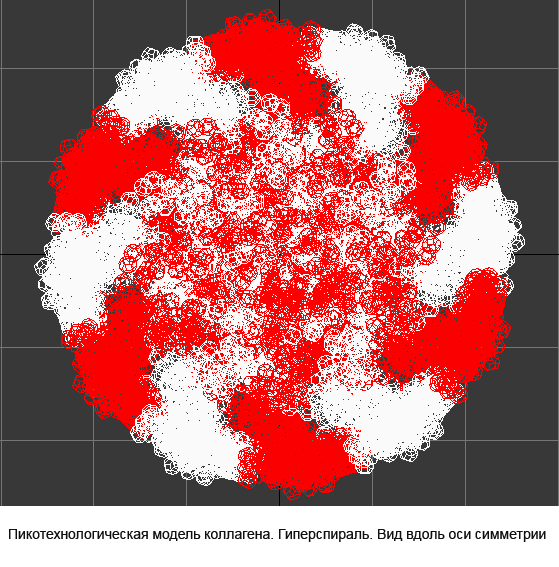
Collagen, 3D structure
Collagen, 4-structure, Helix
Collagen, 4-structure, Helix
Collagen, 4-structure, piko-precision
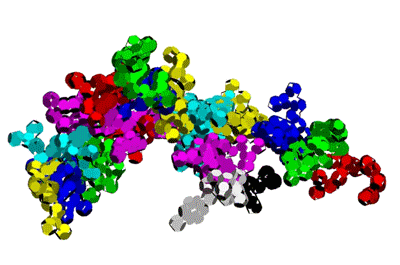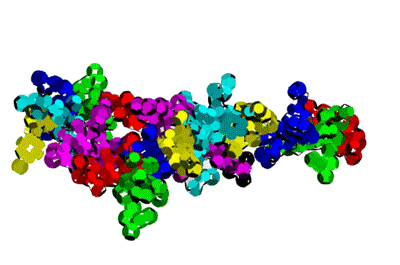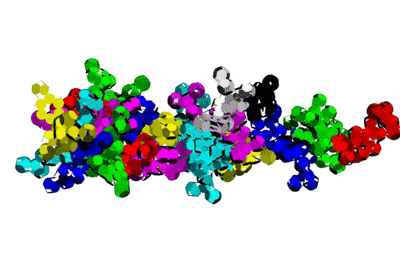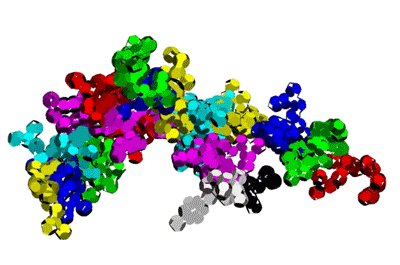
Insulin, 3D-structure
Insulin, 3D-structure Insulin, 2D-structure (PIKO-2D standart (project)) compositional code: 1,1,4,1,1,4,1,4,4,1,4,1,4,1,4,1,1,1,4,1,1,4,1,1,4,4,3,3,4,1,4,1,1,1,1,1,4,1,1,3,1,1,1,1,1,4,1,1,1,4,1 Kushelev's script for
3DS Max: angx=#(-24,180,60,-45,-45); angy=#(-10.89167,83,0,15,15); angz=#(92,120,60,110,110) dx=#(2,1.5,1.6,2,2); dy=#(0.6,1,0.8,1,0.6); dz=#(-0.45,0,-0.6,-0.15,-0.45) kk=#(1,1,4,1,1,4,1,4,4,1,4,1,4,1,4,1,1,1,4,1,1,4,1,1,4,4,3,3,4,1,4,1,1,1,1,1,4,1,1,3,1,1,1,1,1,4,1,1,1,4,1,1,1,1,1,1) peptide=box length:1 width:1 height:1 position:[0,0,-.5] wirecolor:[250,250,250]; Converttomesh
peptide for k = 1 to 56 do( element = Hedra family:1 scalep:100 scaleq:100 scaler:100 radius:1 pos:[0,0,0] p:0.4; Converttomesh element attach peptide element; peptide.pivot = [0,0,0]; move peptide [dx[kk[k]],dy[kk[k]],dz[kk[k]]] rotate peptide angx[kk[k]] [1,0,0]; rotate peptide angy[kk[k]] [0,1,0]; rotate peptide angz[kk[k]] [0,0,1])
Kushelev's script - top model
Insulin, 3D-structure, piko-precision (PIKO-3D standart (project)) Savin-Kushelev's script result Savin-Kushelev's script for 3DS Max (Light):http://nanoworld.narod.ru/EMBLReader023L_20101113.txt
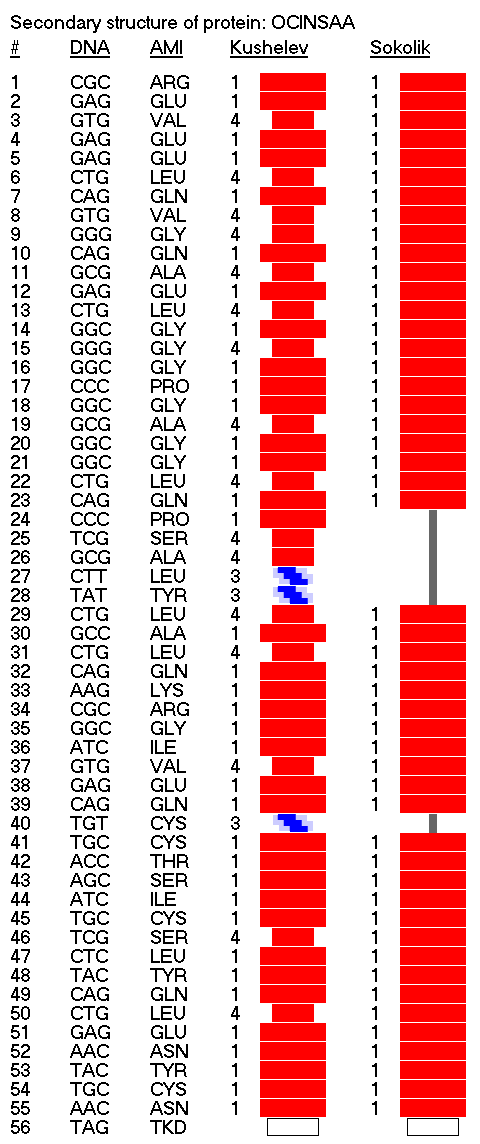
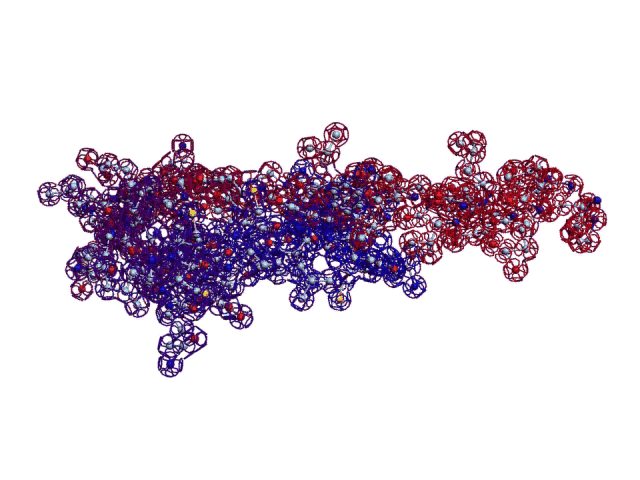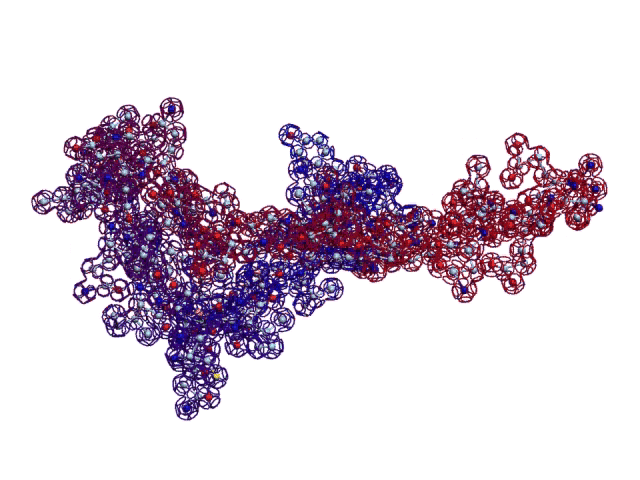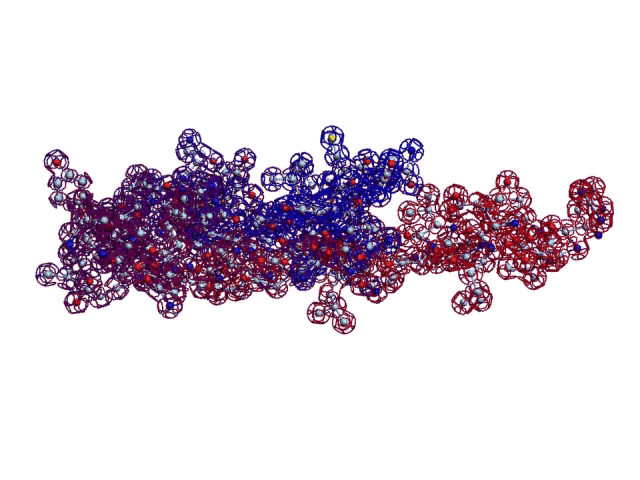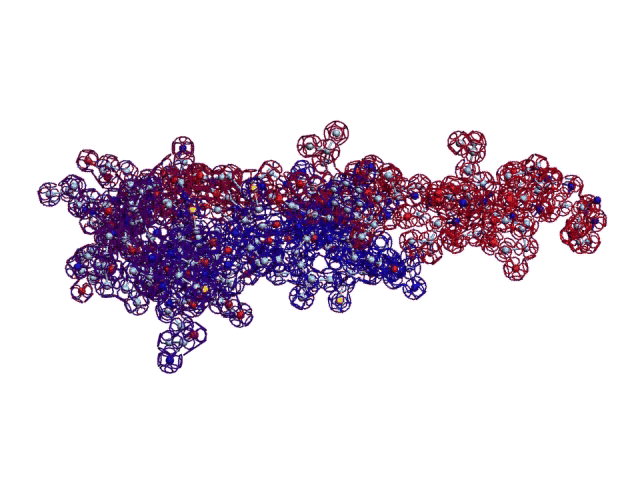
Details: http://nanoworld88.narod.ru/data/212.htm Input data: ID OCINSAA standard; RNA; MAM; 250 BP.
XX
AC M61153;
XX
DT 12-APR-1991 (Rel. 28, Created)
DT 15-APR-1994 (Rel. 39, Last updated, Version 2)
XX
DE Oryctolagus cuniculus insulin mRNA, partial cds.
XX
KW insulin.
XX
OS Oryctolagus cuniculus (rabbit)
OC Eukaryota; Animalia; Metazoa; Chordata; Vertebrata; Mammalia;
OC Theria; Eutheria; Lagomorpha; Leporidae.
XX
RN [1]
RP 1-250
RA Giddings S.J., Carnaghi L.,
Devaskar S.;
RT "Nucleotide sequence of a cDNA encoding rabbit preproinsulin";
RL Unpublished.
XX
CC NCBI gi: 165444
XX
FH Key Location/Qualifiers
FH
FT source 1..250
FT
/organism="Oryctolagus cuniculus"
FT CDS <1..168
FT
/note="NCBI gi: 470321"
FT /product="insulin"
FT /codon_start=1
XX
SQ Sequence 250 BP; 36 A; 105 C; 78 G; 31 T; 0 other;
cgcgaggtgg aggagctgca ggtggggcag gcggagctgg gcgggggccc cggcgcgggc
ggcctgcagc cctcggcgct ttatctggcc ctgcagaagc gcggcatcgt ggagcagtgt
tgcaccagca tctgctcgct ctaccagctg gagaactact gcaactaggg gtgcccccca
cccacccctg cccgcgcccc ccacgccccc cgccctcgcc cccacccaat aaacccctcc
acgcgccccc
// Output data: PFRMAT TS
TARGET T00xx
AUTHOR 4236-1297-6301
REMARK Nanoworld Laboratory
REMARK http://nanoworld.narod.ru
REMARK Predictor remarks. See DEMO:
REMARK http://nanoworld88.narod.ru/data/270.htm
REMARK
REMARK Target sequence (56 acids)
REMARK REVEELQVGQAELGGGPGAGGLQPSALYLALQKRGIVEQCCTSICSLYQLENYCN*
REMARK
METHOD Method description
METHOD The strong correlation dependence
of spatial structure
METHOD of the protein from its nucleotide sequence was
METHOD theoretically predicted by physical modelling,
METHOD experimentally discovered and statistically confirmed.
METHOD In the process of biosynthesis the third nucleotide of
METHOD the codon controls the orientation of the amino acid
METHOD forming the concrete spatial isomer that is the
METHOD conformation of the protein molecule cutting off
METHOD competition ways of the forming of 2D and 3D structures.
METHOD On this base the computer program "Pikotechnology" for
METHOD the prediction of 2D structure of the protins on their
METHOD nucleotide sequence was created.
MODEL 1
PARENT N/A 1
ATOM 1 N ARG A 1 10.504 6.874 4.374 1.00 0.50 A
ATOM 2 CA ARG A 1 9.740 6.273 4.413 1.00 0.50 A
ATOM 3 C ARG A 1
9.285 5.939 3.569 1.00 0.50 A
Details: http://subscribe.ru/archive/science.news.nanoworldnews/201201/13192033.html 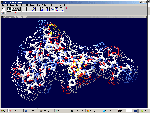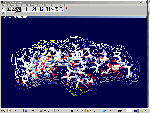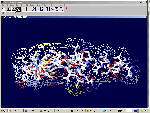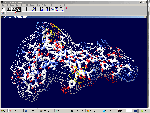
1. Sokolik V.V. Algorithm of protein structural
template decoding according to its determined nucleotide sequence // Fist International Conference “Fundamental Medicine: From Scalpel Toward Genome, Proteome and Lipidome” (April 25-28, 2011), Pax Grid Virtual Conferences, Kazan, Russia. – P. 117-119. 2. Sokolik V.V. Protein is coded in genome and synthesized
in ribosomes as a structural template of a rotameric version sequence of peptide bound configuration // The International Moscow Conference on Computational Molecular Biology, МССМВ-11 (July 21-24, 2011), Moscow, Russia. – P. 347-348.
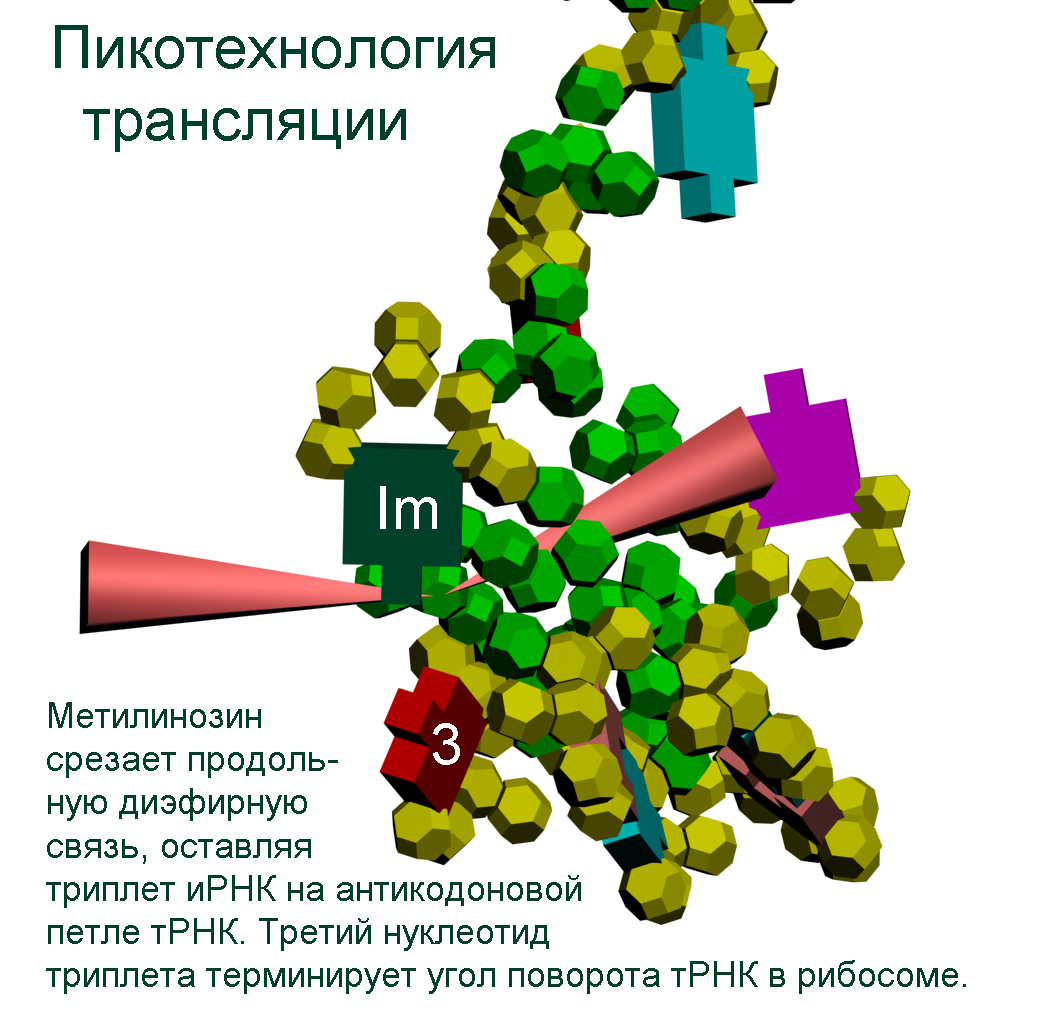
The translation mechanism Механизм трансляции
Pikotechnology (Manuals for schools)
DNA-step model
Nucleotides DNA model
tRNA model
tRNA (detail) model methyl inosine model
ACC-end model
Translation makhanizm 
The translation mechanism 
Details
Rybosome's navigation system Disqussion 

NANOWORLD. The Power Engineering of the Future (popular) SOVIET SCIENCE AND TECHNOLOGY "Soviet Land", #1, 1991, page 8. An account about nanoworld, a sensational discovery of Soviet physicists. *** Fragment of "New Dawn", #1, 1991. I was just a little kid when my father taught me how to ask all sort of questions. "First, you should ask yourself: ask others only if
you don't know the answer". My classmates used to make fun of me, because I always showered teachers with questions. I graduated from high school on top of the class: everybody was quite surprised at the fact that a "dunce" like me had accomplished the seemingly impossible. 
"When I was in my eighth grade my physics teacher Rufina Romanova asked me to hang up over the blackboard a teaching aid (I was the tallest guy in my class) showing the electromagnetic wave scale and the visible light
and ratio wave range. "As distinct from ratio waves, light, which is regarded here as electromagnetic radiation, has the properties of particles. How come?" I asked myself as I climbed up to hang the diagram. I was unable to get an answer to me question back then. The textbooks were rather ambiguous about the whole thing, referring to this property of light as a "wave-particle". I was unable to solve the problem much later, when I was already a freshman at Moscow's power-engineering institute.
Then it dawned on me that a photon might be nothing more than a self-focusing electromagnetic wave package. But this didn't make things easier. "Why do the waves focus at all?"; "At what point do the corpuscular properties begin to manifest themselves?"; "From what substance are light waves formed?"; "Do photons have a structure?"; "If not, then why?" These and other questions seemed mind-boggling... One fine day I happend to be present at a seminar conducted
by Yuri Trusov, working with the Philosophy Institute of the USSR Academy of Sciences, asking him all these questions. Trusov showed me the way the classics tackled different problems. 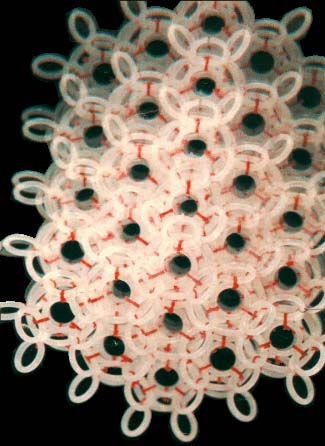
"I used the analogy method to compare the structure of light and sonic waves: this enabled me to modify the mechanical ether model which had been suggested by Maxwell way back in 19th century. The size of ether elements was correlated with the particles that had been theoretically calculated by Max
Planck at the turn of the century (these were called "Planckions" in his honour). The size of these particles is 25 orders of magnitude smaller then that of the atom: that's why, as distinct from atoms, the objects of the microworld, I suggest another name for ether elements, the 'nanoworld' (see photos). My model showing the structure of the nanoworld helped find the answers to the questions that had amassed over the past ten years. These answers are illustrated with photos of models (see cover and text). End. 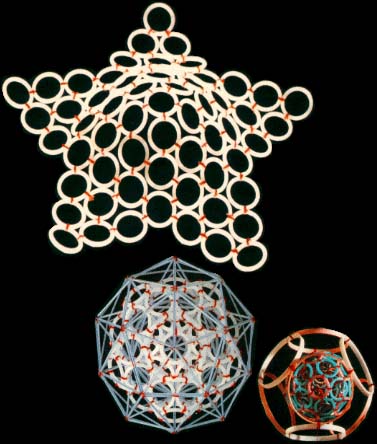
Beginning of the text from "Soviet Land": It all began with question – "Are photons waves or particles?" While trying to provide an answer, Alexander Kushelev propounded the theory that photons are self-focusing electromagnetic waves. But he was immediately faced with two other questions – "Waves of what?" and "How do they become focused?"
Using the standard scientific analogy method for comparing the structures of light and sonic waves, the young genius constructed a model showing the structure of the nanoworld. By the way, in a nanoworld, the linear dimensions of elements are 25 orders of magnitude smaller than the atom proper. The model has helped in providing answers for age-old questions which the physicists have been trying to answer and posed some new ones. Here is what Kushelev, who has been working together with Dmitry Kozhevnikov, has to say: "My model showing the structure of the
nanoworld helped in finding the answers to the questions that had accumulated over the past ten years". This answers are illustrated with the photos of models. 
As far as the model of the nanoworld structure is concerned, the rings shown here symbolise the closed wave vortexes existing in the pikoworld (after the nanoworld, this is yet another
step enabling one to penetrate deep inside matter). When the vortexes oscillate in relation to each other, this corresponds to the propagation of an electromagnetic wave. In that case, the photon is a self-focusing electromagnetic wave, i.e., a ray. If a ray is twisted into a ring, then we will get an electron. In case rings (electrons) gather round an atom, a system of ring-shaped polyhedral electronic atomic shells is formed. So far it is next to impossible to pinpoint the nanoworld's structural elements;
however, TV and radio sets detect their fluctuations almost daily. Models consisting of 1, 2, 8, 18 and 32 rings correspond to stable atom shells and, at the same time, to symmetrical polyhedrons. If such models are made of magnetic rings, then the force of magnetic gravitation will bind them together. If we substitute cog-wheels for the rings, these will start spinning simultaneously; the direction of the spin is going to alternate accordingly. "Atom models join together to form molecule, crystal and quasi-crystal models. 
Rings and ring-shaped polyhedrons may form a fractal, in which the element reproduces its own shape but on an entirely different scale. There are ring models, as well as standard rod models, which complement each other. By the way, the management of the Moscow-based Exhibition of Economic Achievements has awarded us a silver medal for furnishing additional information on inter-atom distances and directions, obtained
by means of ring-shaped polyhedral models. The eight starfish-shaped elements give the human body its shape. The same elements also constitute the building material of animal organs and even cellular structures, down to the shell of the simplest virus. The last model, created while preparing this article, which has been named the DNA model, is in fact a spiral band nine atoms wide and one atom thick. While conducting our research, we "accidentally" managed to solve a 2000-year-old task – that of angular trisection. "Who needs all this?" – an inquisitive
reader might ask. Well, here's what the great Thomas Alva Edison said on this score: "What uses a child might have? He will grow up and become a man". Still I will try and explain the possible practical implications of the nanoworld studies. A rough estimate of energy density inside the spinning vortexes that constitute the structure of the nanoworld shows that one cubic metre of structure contains some 10114 joules of energy. At the same time, one cubic metre of nuclear fuel contains approximately 1018 joules of energy, i.e., 96 orders of
magnitude less. The most important thing is that we can tap this fantastic energy and relay it back to the nanoworld's structure by means of a resonance process, similar to the electron, without having to resort to the rather risky chain reaction. In case resonance conditions are disrupted, the energy flow will be reduced and the possibility of an explosion will be totally ruled out. Surely, one can use this type of energy both for destructive and peaceful purposes. The nanoworld's autonomous energy source may
be used to power a 'flying saucer', which would not have to refuel every now and then. Besides, the nanoworld may well bring about a revolution in power engineering by putting an end to the improvised 'metabolism' of today. However, the construction of a relevant energy transformer may prove a formidable task, indeed, requiring the closest cooperation of all the world's scientists. You see, even in the most primitive type of transformers, the electron, (which, by the way, has lowest energy density), the intensity of the magnetic
and electric fields surpasses that of artificially-induced fields by a million times. Another important thing is that the intensity of oscillations inside an electron reaches hundreds of billions of oscillations per second. So, we cannot use the present-day technological means at our disposal to tap the nanoworld's energy source; it will be the same as using a stone axe to split the atom. The most important thing is not destroy civilisation as we know it. Dmitry Kozhevnikov, whom I met some twelve months after graduating from Moscow State
University, is tackling the same kind of problems. At that time I was working as an engineer with the technical diagnostic lab at an R&D institute. Dmitry was then an under-graduate student, who also took a great deal of interest in the structure of the atomic nuclei. Quite soon he suggested that we make ring-shaped polyhedral internal electronic atom shells. These consisted of eight to 32 electrons, and quite soon it turned out that we had made some major discoveries. I even suggested naming some of these
shells after my friend. The 18-electron shell, for one, enabled us to discover a new geometrical figure – a polyhedron consisting of 18 facets or several regular polygons. I and Dmitry started working on a regular basis, and together we have made a few discoveries in the nanoworld sphere. For example, we have succeeded in deciphering the geometry of various types of chemical bonds. We have also found out why the ring-electrons change their diameters while approaching a nucleus or drifting away from it, etc. We continued working together at the exhibition
of young people's scitech creativity hosted by the Exhibition of Economic Achievements. At present quite a few of our models are displayed there, with each of us contributing an equal number. But, whereas my models mostly deal with the shape of elementary particles, a topic which lies in the realm of physics, a lot of Dmitry's models deal with physical and organic chemistry. Dmitry has taken up structural modelling in the applied with the express purpose of linking it to practical results. This did not seem important to me at first,
but in the long run my friend's work helped in creating models of organic molecules, which in its turn enabled us to create a new DNA model of the matrix type. As I have already said, this model resembles a spiral band one atom thick and nine atoms wide, with the south and north electron fields arranged just like black and
white squares on a chess-board. The DNA band proper owes its peculiar shape to the fact that the phosphorus atoms, spread along the rim of the band, have a greater diameter then the carbon atoms. This is our latest achievement, by the way. *** How Your models correlate with X-ray ?
Construction of the scale model of structure of
a protein 1992 A.Yu. Kushelev,
S.A. Pisarzhevsky
The Nanoworld Laboratory
Scientific-industrial association "Politechnology"
107005, Russia, Moscow, Gospitalniy per., 10,
The Bauman Moscow State Technical University SUMMURY The strong correlation dependence of spatial structure of the protein from its nucleotide sequence was theoretically predicted by physical
modelling, experimentally discovered and statistically confirmed. In the process of biosynthesis the third nucleotide of the codon controls the orientation of the amino acid forming the concrete spatial isomer that is the conformation of the protein molecule cutting off competition ways of the forming of 2D and 3D structures. Keywords: nanoworld, circular model of electron, composition genetical code, prediction of protein structure. The prediction of the spatial structure of the proteins is the important problem of modern molecular biology [1, 2, 3]. The circular model of the electron was created
by us what allows to apply the laws of classical physics to "nonclassical" objects, in particular to create the scale geometrical models of the electron covers of the atoms and the molecules. In our classical models of the microobjects the electrons are presented by the circular magnets. We marked for the simplification of further reasonings that the consideration of the inner structure of the electron, the dependence of the radius of the electron ring from the tension on of the nuclear field and other
physical nuances of the circular model of the electron go out from the limits of this article. MATERIALS AND METHODS The main research method was the physical modeling. It consisted in carrying out model experiments with circular magnets representing the electrons. The number of the circular magnets corresponded to the number of the valent electrons of the molecule. The supplementary surfaces imitating compensation of electrostatic influence of the nuclei of the atoms and the other electrons of the molecule were used in the experiments. Some models were made by the method of the symmetrical
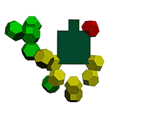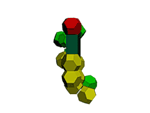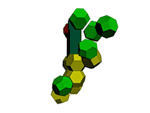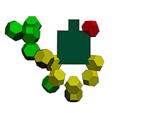
placing of the set number of the rings with due regard for all kinds of the properties of the real electrons. The methods of looking through all the versions, analogy, optimization, systematization and others were used. The data bases PDB and EMBL were used. RESULTS The first experimental result was obtained in the model experiment with the circular magnets which was carried out with the aim of verification of the hypothesis: "The rings-electrons forms the electron cover the polyhedron". Really 8 circular magnets supports the form of the octahedron from the rings packed up on the sphere of the
electrostatic equilibrium of the electrons. This model of electron cover corresponds to the experimentally discovered fermi-surface (fig. 1). The substitution of the four rings-electrons by the unfinished octahedrons (without one ring) turns the form of the octahedron atom to the familiar to us form the tetrahedron molecule. Electron surfaces of the molecules of carbon tetrachloride and phosphoric acid have such a form. We constructed more than 1500 circular models of the various chemical compounds. The special class of chemical compounds in conjugated systems. The model experiment
showed that mutual influence of the similar to carbon atoms appeared to be the cause of the reconstruction of the electron structure of the molecule from polyhedron to polylayer what leads to formation of the "flat" conjugated systems, for example, molecule of benzol. The composition of polyhedron and polylayer atoms and molecules allowed us to compose ringsided structure of DNA nuleotides (fig. 2).The structure of our model of DNA doesn’t contradict generally accepted model. We constructed the model of the group CCON. The different versions
of the mutual arrangement of the groups CCON in the protein structure are given on the figure 3. T-RNA holds amino acid by the ACC-end. The feature of the structure of t-RNA found by us consists in the loop structure of the ACC-end.The last nucleotide of the triplet ACC is turned on the hinge-molecule of the phosphoric acid thus that the process of the formation of the additional diether bond by its free group PO3 with the group PO3 of the complementary chain of RNA has appeared possible. The last from the two nucleotides forming 3’-end of t-RNA
has inversion magnetic properties,which distinguish all nucleotides of the reverse chains of DNA and RNA from the nucleotides of the straight chains. The northern magnetic poles of the two electrons (one from oxygen and the second from nitrogen),by which the cytosine -N ends ( in which the northern poles of the electrons forming hydrogen bonds with the complementary base are inverted outside) and the southern magnetic poles of the two electrons of the cytosine-S (in which the southern poles of the electrons forming hydrogen
bonds with the complementary base are inverted outside) are disposed complementary to the four electrons of the peptide group,which oriented by the group of nitrogen along the rotation axis of t-RNA.The amino acid may be placed by the manipulator-t-RNA in the growing protein chain with the different angle of the turn around the rotation axis.The angle of the turn is regulated by the varible loop of t-RNA and the distance between the third mucleitide of the codon of m-RNA and the first nucleotide of the anticodon
of t-RNA.The distance between the first nucleotide of the anticodon (inosine) and the third nucleotide of the codon depends of the nature of the base complementary for inosine (A, C, G, T). We have constructed the circularside models of the proteins with the known structure in particular myoglobin, insulin, troponin, oxytocin. The sorting out helped to choose version of the connection of the models of group CCON, which gave the forms of these proteins. It turned out that ,for example, the amino acid glycin is encoded by the code
GGA if it was a member of beta-layer, by the code GGG if it was a member of 3/10 alpha-helix where hydrogen bond wasn’t with the forth but with the third amino acid residue and finally by the code GGT if it was the second amino acid residue of the beta-turning. Thus the composition genetical code was composed showing how the amino acid is disposed relatively previous one depending on its genetical code(table 1). The regular repetition of the version 1 leads to the formation of alpha-helix. The repetition of the version 2 leads to the formation of beta-layer.
The turn of the chain in beta-layer demands the sequence of the versions 1-4-1. The repetition of the versions 3 leads to the formation of xi-helix deviding alpha and beta parts of the proteins. The repetition of the version 4 leads to the formation of the diminished alph-helix.Its structure differs from the structure of the ordinary alpha-helix :the hydrogen dond between the group CO and the nitrogen group is closed not on the fourth but on the third amino acid residue. The special case is the helix of collagen. The versions 1, 2, 3, 4 is alternate in it. Collagen has
the unusual structure.Its peculiarity is the presence of the soft axis of the symmetry rolled up into helix. This helix has elastic properties. The geometry of the amino acids and the table of the composition genetical code we layed to the foundation of the program "Pikotechnology", which assemble the structure of the protein molecule on the screen of the PC. Had used accessible for us codes the program reckoned 30 varieties of keratins to the alpha-structural proteins and 3 types of fibroins to beta-structural proteins. We compared the data about
2D structure of the proteins from the Brookhaven Protein Data Bank with the data obtained with the help of our program by the codes of the same proteins from Oxford Bank of Nucleotide Sequences. Eight corresponsing sequences from the Oxford Bank were found for 314 structures of the Brookhavon Bank.Per cent of the content of alpha-codons corresponding to the alpha-helix parts (according to X-ray analysis) is indicated below. Cholesterol oxidase (1.1.3.6) 92.43 Arabinose-binding protein 57.52 Lysozyme (3.2.1.17) 41.54 Aldolase (4.1.2.13) 73.00 Aminotransferase (2.6.1.1) 72.02 Trp-repressor (E-coli) 61.68 Glycogen phosphorylase (2.4.1.1) 85.75 Myoglobin 70.59 The data obtained by us demands the revision of the existing views on the structures
of the proteins, in particular on interpretation of the data of the X-ray structural analysis. The fact is that many globular proteins according to our model experiments consist in sewed together alpha-helix strips, which is interpreted as beta-strips now. DISCUSSION The growing polypeptide chain is synthesized deeply inside the ribosome. However every aminoacid may get the conformation of the extended beta structure (in this case it is encoded by beta codon) and alpha structure (in this case it is encoded by alpha codon). The diameter of the
alpha structure practically does not differ from the diameter of the extended beta structure. Moreover, the diameter of the structure depends on the length of the radicals which are included in the composition of the alpha and beta structures more strongly. For example, the replacement of the radicals of Gly by the radicals of Trp enlarges the diameter of beta and alpha structures approximately in three times, while the diameter of the alpha helix exceeds the diameter of beta structure less than in 1,5 times. Except the extended beta
structure (in our model it corresponds to the version 2, for example, for Gly it is the triplet gga) in the channel of the ribosome the alpha helix (version 1, for example, for Gly it is the triplet ggc), 3/10 alpha helix (the version 4, for example, for Gly it is the triplet ggt) and at last the so called xi structure (the version 3, for example, for Gly it is the triplet ggg) may be placed. More than 20 residues forming one of these four structures really should be incorporated before the chain quits from the channel in the ribosome.
However the nature of the codon of any residue may influence its conformation in the protein structure forming either the site of extended beta structure or the site of alpha helix or the site of 3/10 alpha helix or the site of xi structure, which a little differ on diameter in comparison with the length of the largest radicals. Comparing structures of the proteins received with the help of this program with the data of X-ray structural analysis the specialists engaged in study of the protein structure discover divergences consisting
in displacing of the ends of alpha-helix at time even for five aminoacid residues. Can X-ray analysis make such a mistake? Let's reason. What is alpha-helix? This is the helix each coil of which includes approximately four aminoacid residues. Consequently residues with numbers 1 and 4 are located side by side and are binded by hydrogen bond. Adjacent residues to the residue 4 (3 and 5) are also located very closely from the residue number 1 (to say not on 1.5 A but on 2 A). It is turning out that making a mistake on 1 – 1.5 residues we may actually make a mistake on 5 residues. So if we see that our program shows all turning
of protein alpha-helix with "the mistakes up to 5 aminoacid residues" then the conclusion can be made that it works faultlessly, but X-ray making a mistake on one and a half residues may make a mistake on 5 residues. We would like to supplement the article by the demonstration of the serviceability of our method. For demonsrating we have selected the fragment of lysozyme, which structure is investigated by different metods. Indisputable in its structure is S-S link between amino acid residues Cys96 and Cys117. Our program "Pikotechnology"
generates 3D structure of the protein autamatically, utilizing the table of the forms of the amino acid residues, the table of composition genetical code and genetical code from the EMBL-file. Having obtained the coordinates of the atoms with the help of our program we see, that the site of the protein Cys96…Cys117 really forms the cycle througth S-S link. REFERENCES [1] D.J. Wales, H.A.Scheraga. Global optimization of clusters, crystals, and biomolecules. Science, 285(5432) (1999), 1368-1372 [2] D. Shortle. Structure prediction: The state of the art. Curr Biol, 9(6) (1999), R205-209 [3] S.Takada. Go-ing for
the prediction of protein folding mechanisms. Proc. Natl. Acad. Sc. USA, 96(21) (1999), 11698-11700. THE TABLE OF COMPOSITON GENETICAL CODE
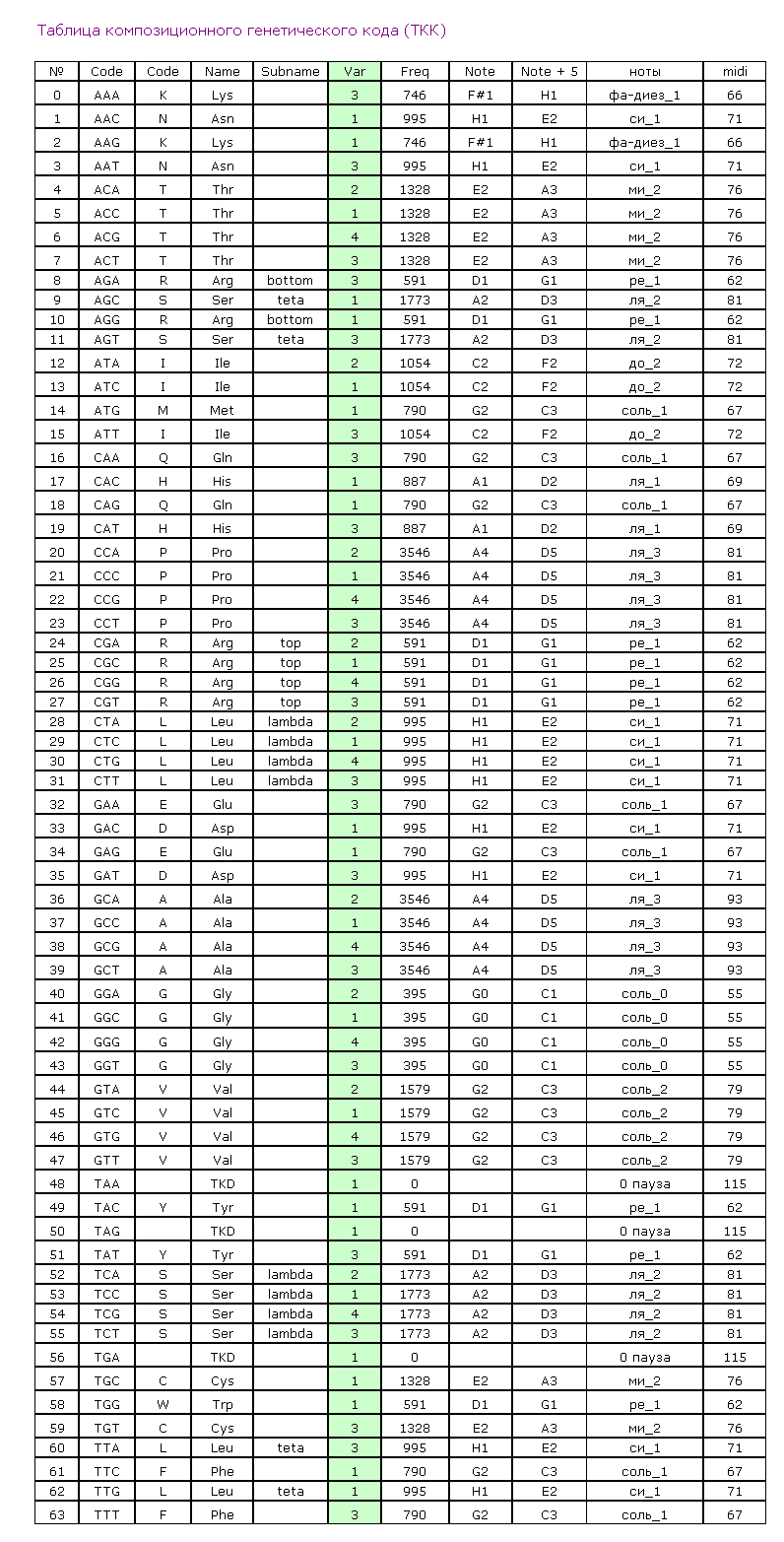
The version 1 corresponds to the entry of the residue to the 4/10 alpha-helix. The version 2 corresponds to the entry of the residue to the classical beta-layer. The version 3 corresponds to the entry of the residue to the beta-turn. The version 4 corresponds to the entry of the residue to the
3/10 alpha-helix. FIGURE LEGENDS Figure
1: The structure
of the 8-electron cover for which the majority of the atoms of the mendeleyev table are completed. Figure 2: The structure of the nuckeotides. Figure 3: The model of the superposition of the group CCON with the previous group CCON in accordance with composition genetical code. Version 1: the point F coincides with the point B (oved counter clock-wise for some degrees). The version 2: the point F coincides with the point A. The version 3: the point F coincides with the point C. The version 4: the point F coincides with the point B (moved clock-wise for some degrees). Figure 4: The structure of the fragment of lysozyme created by the program "Pikotechnology". 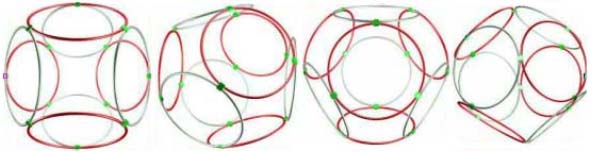
Fig. 1
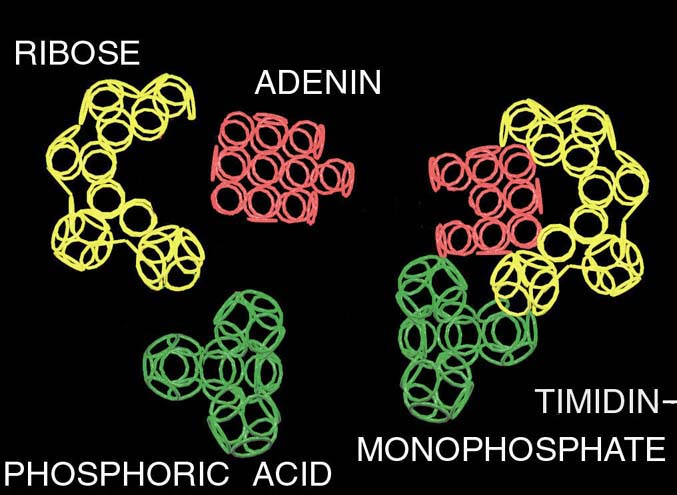
Fig. 2
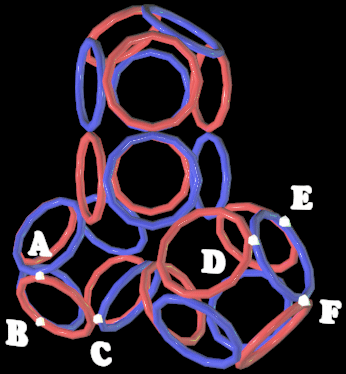
Fig. 3
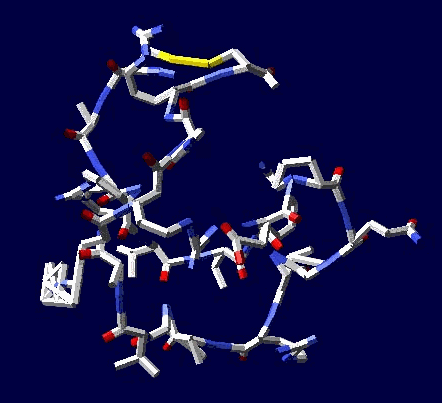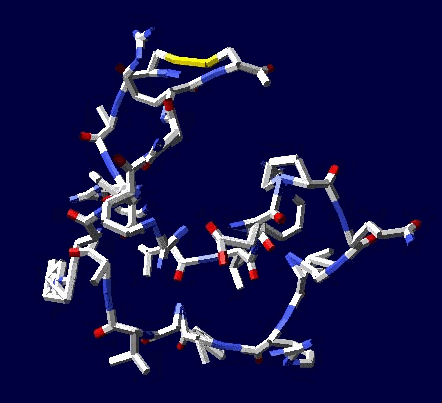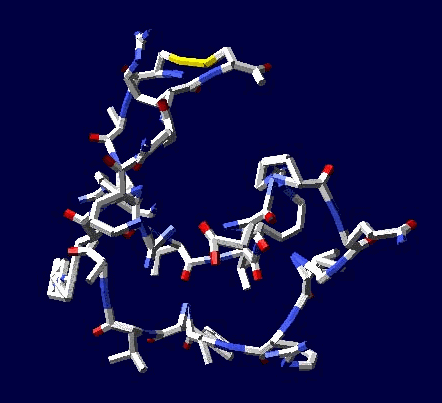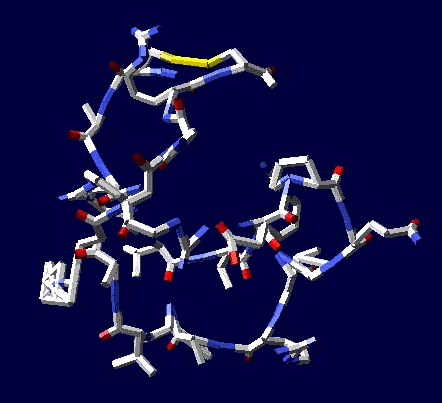
Fig. 4 Prof: Why you offer the new standard? The PDB standard can't be modified? To add precision etc. Кушелев: У нас есть инструмент, имеющий точность пикометр. Этот инструмент показал систематическую ошибку РСА. Попытаемся понять, какова точность РСА. Я решил проверить результаты программы Пикотех с помощью РСА. При этом я обнаружил странную закономерность. Спиральные участки, обнаруженные с помощью программы Пикотех совпали с данными РСА,
но со сдвигом до 5 аминокислотных остатков. Если бы алгоритм был ошибочным, то свиг был бы произвольным. Когда же свдиг не превышает 5 остатков, а контур макромолекулы совпадает, становится понятно, что это - ошибка РСА. We have piko-precision system. This system show systematic X-ray defects ... Let's understand X-ray precision. I have decided to check up pikotech by X-ray. I have found out strange law ... Helix sites coincide with shift 0... 5 amino acid residues. The wrong algorithm would increase divergences with X-ray data
has opened unknown earlier regular errors. Кушелев: Пикотехнология вскрыла неизвестные ранее систематические погрешности рентгеноструктурного анализа. Рентген ошибается на концах спиральных участков до 5 аминокислотных остктков (amino acid residues). Если бы алгоритм пикотехнологии был ошибочен, то был бы не сдвиг границ спиральных участоков, а полное несовпадение. Позднее выяснилось, что РСА (X-ray) не замечает разрывов спиральных участков, не отличает альфа-спираль от 310-спирали. Большинство белков, где считалось присутствуют
протяжённые бета-слои, оказались гнутыми спиралями, т.е. чередованием альфа-спиральных участков и 310-спиральных. Pikotechnology show unknown earlier regular X-ray errors. The X-ray is mistaken on the ends helix Sites to 5 amino acid residues. If the algorithm Pikotech was erroneous, there would be not a shift of borders Helix sites, and full discrepancy. Later it was found out that X-ray doesn't notice ruptures of helix sites, the alpha-helix doesn't distinguish from 310-helix. The majority of peptides where was considered are
present Extended beta layers, have appeared bent helix, i.e. alternation Alpha-helix sites and 310-helix. 
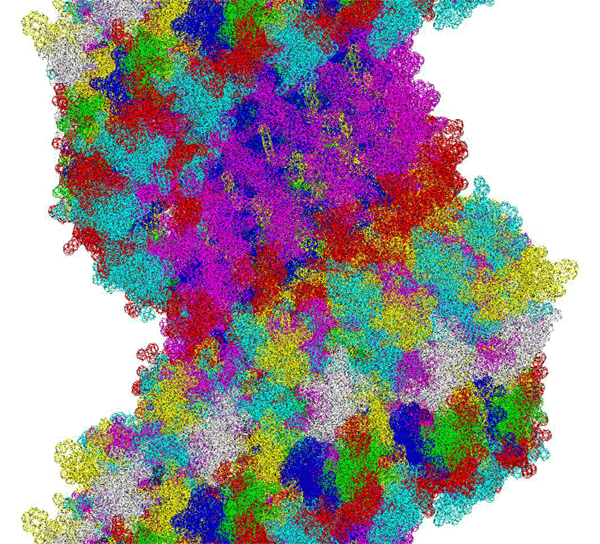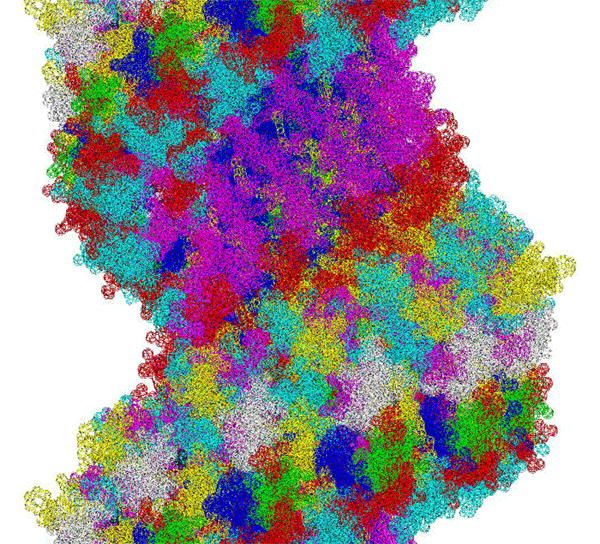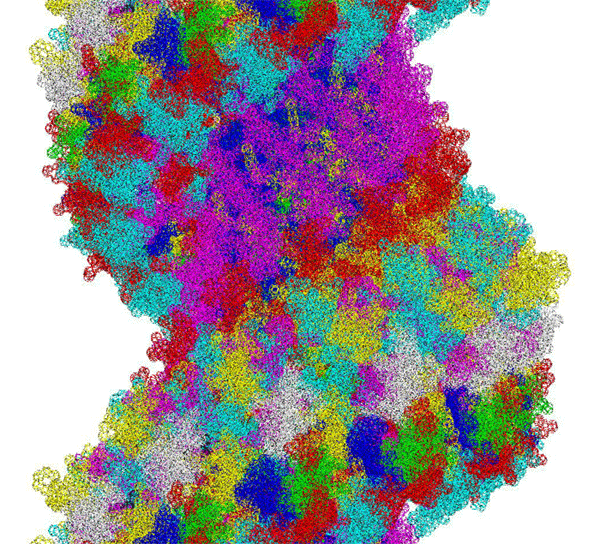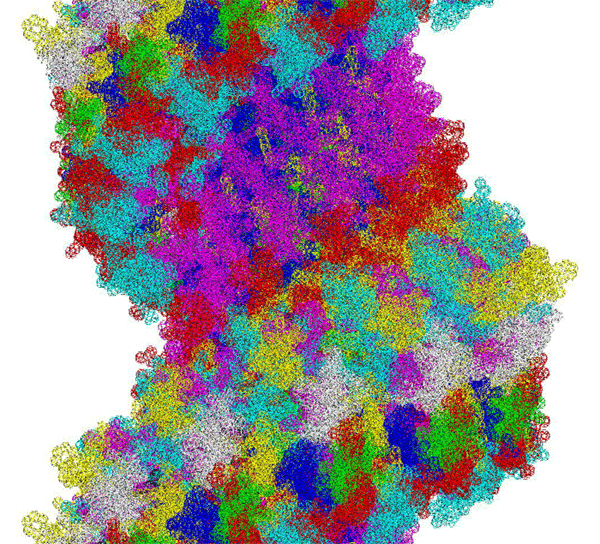
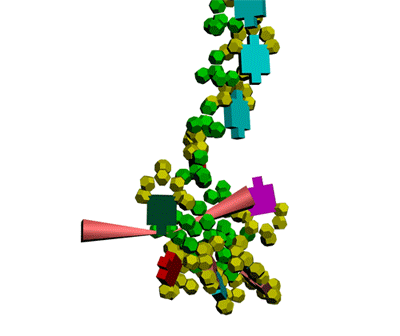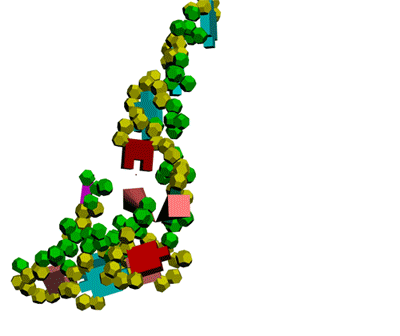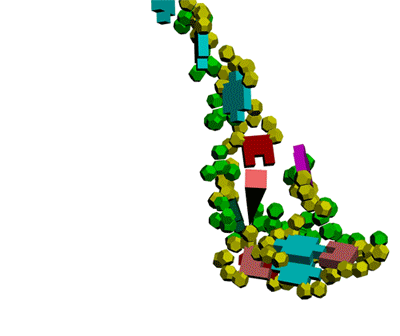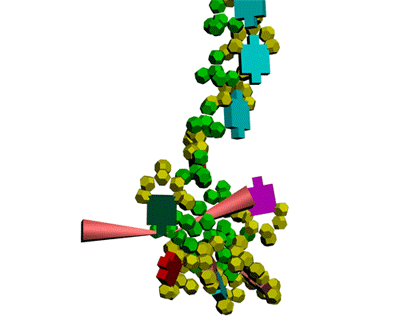
This alpha carbon backbone model of insulin, a 51-amino acid peptide hormone, features 2 chains (A is orange and B is purple) joined by two disulfi de bonds. This model is useful in teaching basic principles of protein structure – including quaternary structure. The insulin model can also be used in conjunction with the Insulin mRNA to Protein
Kit (above). Mini Models are made of plaster by rapid prototyping and should be handled with care. Meets National Science Education Standards.
Kushelev: Let's compare to the structure of insulin received by Pikoteh: 
Disqussion / Обсуждение
Вести c ускорителей ... News from accelerators... 
Резонаторы прибыли на ускоритель "B". Эксперимент планируется во второй половине января. Материал с форума лаборатории Наномир:Георгий Шишков: Правильно ли я понимаю, что будильники будут в вакуумированной камере? Если так,
то заниматься подрегулировкой положения резонаторов относительно луча - неудобно. Если из-за этого возникнут затруднения, предложу схему позиционера с пневматическим управлением (если надо). Но будем надеятся, что и так все получится ... Кушелев: Нет. Луч из ускорителя идёт в атмосфере. Auto
translation: George Shishkov: whether Correctly I understand, what alarm clocks will be in vacuum to the chamber? If so to be occupied by readjusting of position of resonators concerning a beam - it is inconvenient. If because of it there will be difficulties, will offer the scheme position air-controlled (if it is necessary). But we will be belive as so all will turn out... Kushelev: No. The beam from the accelerator goes in atmosphere. Обсуждение.
Гомологический критерий Кушелева В настоящее время биологи пытаются моделировать структуру неизученных белков методом гомологии, т.е. совпадения аминокислотных последовательностей. В этом деле им может помочь гомологический критерий Кушелева. Нужно определить композиционный код предполагаемых гомологов. Если он совпадает, значит перед нами
действительно гомологи. Если не совпадает, то формы белков могут существенно отличаться. Предлагаю испытать гомологический критерий Кушелева на известных гомологах. Композиционный код белка легко получить по кодирующей его нуклеотидной последовательности с помощью программы от Prosolver:http://nanoworld.narod.ru/Pikotech2D_3.6.zip В версии 2011 года композиционный
код можно скопировать в буфер обмена данными Windows и сохранить в текстовом формате. Так выглядит композиционный код инсулина: 1,1,4,1,1,4,1,4,4,1,4,1,4,1,4,1,1,1,4,1,1,4,1,1,4,4,3,3,4,1,4,1,1,1,1,1,4,1,1,3,1,1,1,1,1,4,1,1,1,4,1 Kushelev's homological criterion Auto translation: Now biologists try to model structure of not studied peptides a homology method, i.e. coincidence amino acids sequences. With this business they can be helped by Kushelev's homological criterion. It
is necessary to define a composite code of prospective homologs. If it coincides, means before us really homologs. If doesn't coincide, forms of peptides can essentially differ. I suggest to test Kushelev's homological criterion on known homologs. Composite code of peptide it is easy to receive on coding it DNA-sequences by means of the program from Prosolver: http://nanoworld.narod.ru/Pikotech2D_3.6.zip In version 2011 the composite code can be copied in a clipboard data Windows and to keep
in a text format. So the composite code of insulin looks: 1,1,4,1,1,4,1,4,4,1,4,1,4,1,4,1,1,1,4,1,1,4,1,1,4,4,3,3,4,1,4,1,1,1,1,1,4,1,1,3,1,1,1,1,1,4,1,1,1,4,1 Victoria пишет:Всё верно  . .
Я о надуманности гомологии белков одного семейства писала здесь: Victoria writes: All is true. I about artificiality of a homology of peptides of one family wrote here: 1. Соколик В.В. Пространственная структура гомологов основного актина и α-актина 1 различна / Сборник материалов I Международной научно-практической конференции «Наука и современность – 2010» в 3-х час-тях / Под общ. ред. С.С. Чернова - Новосибирск: «СИБПРИНТ», 2010. – 278 с. Ч. 1, С. 41-46.
и здесь:
2. Соколик В.В., Мандзюк Т.С. Структурные механизмы агрегации синуклеина в возрастной патологии конформации белка / Материалы IX Междуна-родного симпозиума «Биологические меха-низмы старения». (26-29 мая
2010 г.), Харьков. – С. 20-21.
Обсуждение / Discussion
Приглашение к сотрудничеству для людей умеющих самостоятельно мыслить; не просто умных, а мудрых, которые чувствуют, где истина
Лаборатория Наномир готова к любому взаимовыгодному сотрудничеству. У нас есть сторонники как явные, которые помогают морально и материально, есть очень много пассивных наблюдателей, есть и ярые противники, которые используют
любые методы и средства (аморальные и просто преступные), чтобы уничтожить работу лаборатории и дискредитировать ее. В одиночку внедрить технологии, выводящие цивилизацию на новый уровень, невозможно. Благодаря поддержке множества заинтересованных людей проделана огромная работа. Ознакомиться с её результатами можно изучив материал рассылки "Новости лаборатории Наномир". Люди науки могут изучить научные
труды.
Вклад каждого не останется незамеченным в случае успеха в реализации научных проектов. Результаты совместной деятельности принадлежат участникам проекта пропорционально коэффициентам творческого и финансового участия. В этом году были куплены рубиновые шарики для эксперимента на сумму ~1000 долл. В результате было сделано научное открытие, проверена защита диэлектрических резонаторов от перенапряжения. В этом же году, вероятно, можно будет создать микроволновую энергетику,
т.к. удалось найти сырьё (рубин #8), из которого сделаны рубиновые шарики для эксперимента в Дубне. 28 сентября начался эксперимент по созданию "эликсира вечной молодости". Благодаря первому взносу (в размере 500 долларов) Золдракса и поддержке других соинвесторов. Продолжаются переговоры с потенциальными инвесторами по поводу финансирования этого проекта. Созданы первые версии пикотехнологии, с помощью которой Александр Кушелев и Виктория Соколик сделали более10 научных открытий.
Сотрудничество
может быть различным: - участие в научных дискуссиях на форуме (конструктивное) - совместное создание коммерческого продукта - поиск инвесторов - выступить менеджером по продаже готовых коммерческих продуктов - конструктивные предложения по продвижению идей лаборатории Наномир - содействие в проведении экспериментов и т.п.
- написание совместных научных статей и т.п. - материальный вклад (денежный или обеспечение оборудованием и материалами)
Пожалуйста, сообщайте о своем вкладе, чтобы мы зачли Вас как партнера лаборатории Наномир. +7-926-5101703 begin_of_the_skype_highlighting +7-926-5101703 end_of_the_skype_highlighting, +7-903-2003424 begin_of_the_skype_highlighting +7-903-2003424 end_of_the_skype_highlighting, +7-916-8265031 begin_of_the_skype_highlighting +7-916-8265031 end_of_the_skype_highlighting, Skype: Kushelev2009, mail: kushelev2011@yandex.ru
веб-мани: WM-кошелек R426964799301
|












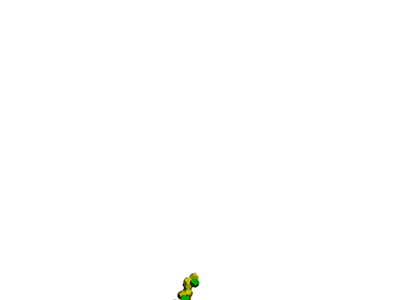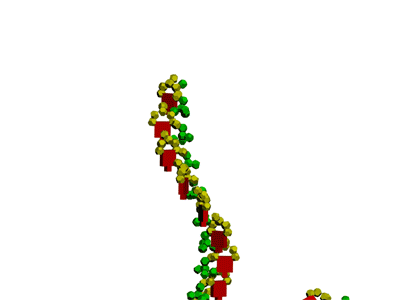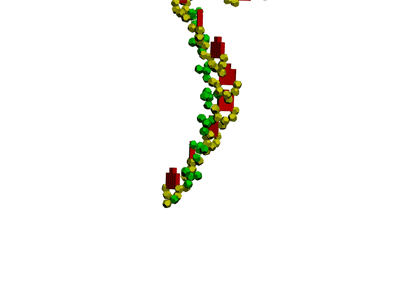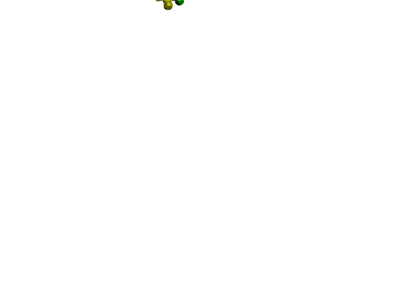



{kind=link}